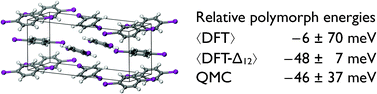
We show that the quality of density functional theory (DFT) predictions for the relative stabilities of polymorphs of crystalline para-diiodobenzene (PDIB) is dramatically improved through a simple two-body correction using wavefunction-based electronic structure theory. PDIB has two stable polymorphs under ambient conditions, and like Hongo et al. [J. Phys. Chem. Lett., 1, 1789 (2010)] we find that DFT makes wildly variable predictions of the relative stabilities, depending on the approximate functional used. The two-body corrected scheme, using Grimme's spin-scaled variant of second-order Møller–Plesset perturbation theory and any of the tested density functionals, predicts the α-polymorph to be more stable, consistent with experiment, and produces a relative stability that agrees with the benchmark quantum Monte-Carlo results of Hongo et al. within statistical uncertainty.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?