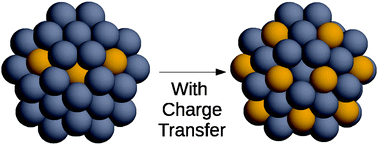
The determination of optimal chemical ordering in nanoalloys, i.e. of the most stable pattern in which atoms are arranged in bi- or multicomponent metallic clusters, is quite complex due to the enormous number of different possible configurations. This problem is very difficult to tackle by first-principle methods except for very small systems. On the other hand, the treatment at the atomistic potential level is complicated in many cases (such as AgAu) by charge transfer effects between atoms of different species in different coordination environments. Here an empirical atomistic model is developed to take into account such effects. The model is used to determine the optimal chemical ordering in AgAu nanoalloys. Charge transfer between atoms is taken into account by a modification of the charge equilibration method of Goddard and Rappé [J. Phys. Chem., 1991, 95, 3358], in which a coordination-dependent electronegativity and hardness are introduced. The model is applied to the determination of chemical ordering in AgAu nanoalloys. It is shown that the inclusion of charge transfer effects is important for improving the agreement of the atomistic model with density-functional calculations, leading to the determination of lower-energy chemical ordering patterns.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


