
Combined Monte Carlo and quantum mechanics study of the solvatochromism of phenol in water. The origin of the blue shift of the lowest π–π* transition
Abstract
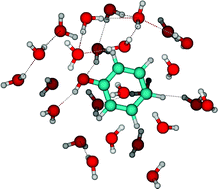
A combined and sequential use of Monte Carlo simulations and quantum mechanical calculations is made to analyze the spectral shift of the lowest π–π* transition of
 Please wait while we load your content...
Please wait while we load your content...

