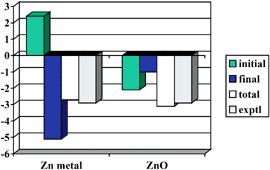
The Zn 2s and 2p core level binding energies of ZnO and a few Zn oxo compounds containing Zn in its oxidation state +2 were calculated by means of wave function based quantum chemical ab initio methods. The computations were performed at two levels of approximation. First, Hartree–Fock calculations were carried out for the ground state of the neutral systems yielding the “initial state” effects, i.e. the shifts of the core level binding energies due to the changes in the chemical environment of the Zn atom under consideration (Koopmans’ theorem level, KT). In the second step, Hartree–Fock calculations were performed for the core ionized states in order to account for the relaxation effects after ionization, i.e. for the “final state” effects (ΔSCF level). Scalar relativistic corrections and spin–orbit coupling were included in a “spin–orbit-coupling configuration interaction” (SOC-CI) treatment both at the KT and ΔSCF levels. In all Zn oxo compounds (Zn4O(formate)6, Zn4O(acetate)6 and several ZnO cubanes) small negative initial state shifts between −1.0 and 0.0 eV (relative to the free Zn atom) were found which are caused by the negative charges at the surrounding O atoms. The relaxation effects vary between −1.0 and −0.5 eV, such that the calculated total shifts are moderately negative (−1.5 to −0.5 eV). Embedded ZnO clusters of increasing size, ranging from Zn13O4 to Zn69O38, were used as models for bulk ZnO, the Zn 2s and 2p core level shifts calculated for these clusters being extrapolated to infinite cluster size. The calculations show that bulk ZnO has a rather large negative initial state shift of −2.1 ± 0.1 eV, due to the Madelung potential at the Zn atom, and a comparatively small relaxation contribution of −1.0 ± 0.1 eV. This yields a total shift of −3.1 ± 0.2 eV (both for 2s and 2p, relative to atomic Zn), which is in very good agreement with experiment, −2.9 ± 0.2 eV. The surprising experimental observation that the Zn 2s and 2p XPS peak positions are nearly identical in Zn metal and ZnO is explained by the fact that the sum of initial and final state effects is accidentally the same for the two systems though the individual contributions differ quite significantly: the initial and final state shifts amount to +2.4 and −5.1 eV for Zn metal vs.
−2.1 and −1.0 eV for ZnO.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


