
DFT plane wave calculations of the atomic and electronic structure of LaMnO3 (001) surface
Abstract
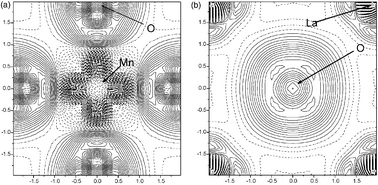
We present the results of ab initio DFT plane wave periodic structure calculations of the LaMnO3 (001) surface. The effects related to three different kinds of pseudopotentials, the slab thickness, magnetic ordering, and surface relaxation are studied and discussed. The antiferromagnetic surface lowest in energy (that is, the spins on Mn ions are parallel in basal plane and antiparallel from plane to plane) has a considerable atomic relaxation up to the fourth plane from the surface. The calculated (Bader) effective charges and the electronic density maps demonstrate a considerable reduction of the Mn atom ionicity on the surface accompanied by a covalent contribution to the Mn–O bonding.
 Please wait while we load your content...
Please wait while we load your content...

