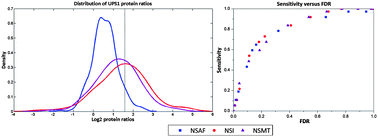
Although widely applied in the label-free quantification of proteomics, spectral count (SC)-based abundance measurements suffer from the narrow dynamic range of attainable ratios, leading to the serious underestimation of true protein abundance fold changes, especially when studying biological samples that exhibit very large fold changes in protein expression. MS/MS fragment ion intensity, as an alternative to SC, has recently gained acceptance as the abundance feature of protein in label-free proteomic studies. Herein, we implemented two formats of MS/MS fragment ion intensity, Spectral Index (SI) and Summed MS/MS TIC (SMT), to alleviate this particular deficiency arising from SC. Both were in forms of replacing SC in the Normalized Spectral Abundance Factor (NSAF) formula, resulting in two algorithms, abbreviated as NSI and NSMT, respectively. The necessity of the normalization process was validated using a publicly available dataset. Furthermore, when applied to another well characterized benchmark dataset, both NSI and NSMT showed improved overall accuracy over NSAF for the relative quantification of proteomes. Hereinto, NSI enabled the sensitive detection of differentially expressed proteins, while NSMT ensured accurate calculation for protein abundance fold change. Therefore, the selective use of both algorithms might facilitate the screening and quantification of potential biomarkers on the proteome scale.

 Please wait while we load your content...
Please wait while we load your content...

