
Substituent effects on aromatic interactions in water†

Abstract
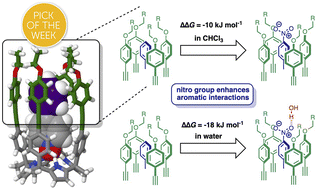
Molecular recognition in water involves contributions due to polar functional group interactions, partial desolvation of polar and non-polar surfaces and changes in conformational flexibility, presenting a challenge for rational design and interpretation of supramolecular behaviour. Conformationally well-defined supramolecular complexes that can be studied in both water and non-polar solvents provide a platform for disentangling these contributions. Here 1 : 1 complexes formed between four different calix[4]pyrrole receptors and thirteen different pyridine N-oxide guests have been used to dissect the factors that govern substituent effects on aromatic interactions in water. H-bonding interactions between the receptor pyrrole donors and the guest N-oxide acceptor at one end of the complex lock the geometrical arrangement of a cluster of aromatic interactions at the other end of the complex, so that a phenyl group on the guest makes two edge-to-face and two stacking interactions with the four aromatic side-walls of the receptor. The thermodynamic contribution of these aromatic interactions to the overall stability of the complex was quantified by chemical double mutant cycles using isothermal titration calorimetry and 1H NMR competition experiments. Aromatic interactions between the receptor and a phenyl group on the guest stabilise the complex by a factor of 1000, and addition of substituents to the guest phenyl group further stabilises the complex by an additional factor of up to 1000. When a nitro substituent is present on the guest phenyl group, the complex has a sub-picomolar dissociation constant (370 fM). The remarkable substituent effects observed in water for these complexes can be rationalised by comparison with the magnitude of the corresponding substituent effects measured in chloroform. In chloroform, the double mutant cycle free energy measurements of the aromatic interactions correlate well with the substituent Hammett parameters. Electron-withdrawing substituents increase the strength of the interactions by a factor of up to 20, highlighting the role of electrostatics in stabilising both the edge-to-face and stacking interactions. The enhanced substituent effects observed in water are due to entropic contributions associated with the desolvation of hydrophobic surfaces on the substituents. The flexible alkyl chains that line the open end of the binding site assist the desolvation of the non-polar π-surfaces of polar substituents, like nitro, but at the same time allow water to interact with the polar H-bond acceptor sites on the substituent. This flexibility allows polar substituents to maximise non-polar interactions with the receptor and polar interactions with the solvent, leading to remarkably high binding affinities.
- This article is part of the themed collections: 2023 Chemical Science HOT Article Collection, 2023 ChemSci Pick of the Week Collection and Emerging Frontiers in Aromaticity
 Please wait while we load your content...
Please wait while we load your content...

