
Forecasting molecular dynamics energetics of polymers in solution from supervised machine learning†

Abstract
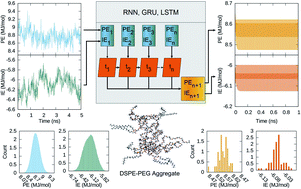
Machine learning techniques including neural networks are popular tools for chemical, physical and materials applications searching for viable alternative methods in the analysis of structure and energetics of systems ranging from crystals to biomolecules. Efforts are less abundant for prediction of kinetics and dynamics. Here we explore the ability of three well established recurrent neural network architectures for reproducing and forecasting the energetics of a liquid solution of ethyl acetate containing a macromolecular polymer–lipid aggregate at ambient conditions. Data models from three recurrent neural networks, ERNN, LSTM and GRU, are trained and tested on half million points time series of the macromolecular aggregate potential energy and its interaction energy with the solvent obtained from molecular dynamics simulations. Our exhaustive analyses convey that the recurrent neural network architectures investigated generate data models that reproduce excellently the time series although their capability of yielding short or long term energetics forecasts with expected statistical distributions of the time points is limited. We propose an in silico protocol by extracting time patterns of the original series and utilizing these patterns to create an ensemble of artificial network models trained on an ensemble of time series seeded by the additional time patters. The energetics forecast improve, predicting a band of forecasted time series with a spread of values consistent with the molecular dynamics energy fluctuations span. Although the distribution of points from the band of energy forecasts is not optimal, the proposed in silico protocol provides useful estimates of the solvated macromolecular aggregate fate. Given the growing application of artificial networks in materials design, the data-based protocol presented here expands the realm of science areas where supervised machine learning serves as a decision making tool aiding the simulation practitioner to assess when long simulations are worth to be continued.
 Please wait while we load your content...
Please wait while we load your content...

