
Molecular dynamics simulations of mono-tethered particles at solid surfaces
 *a
and
Małgorzata
Borówko
a
*a
and
Małgorzata
Borówko
a
Abstract
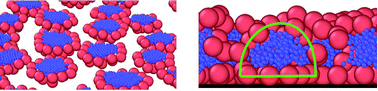
We use molecular dynamics simulations to study the behavior of mono-tethered nanoparticles on solid surfaces. In our model particle–particle and particle–chain interactions are repulsive, while chain–chain interactions are attractive. Two surfaces are considered: the first one attracts particles and the other attracts chains. Excess adsorption isotherms are presented for both the surfaces and different lengths of tethers. The mechanism of adsorption is discussed. We find that depending on the assumed parameters the mono-tethered particles can be adsorbed as single particles or as different aggregates. Our main goal is to explore the structure of surface films. We show that the morphology of the adsorbed layer depends mainly on the type of the surface but the influence of the particle diameter, the chain length and the density is also important. We prove that the shape of aggregates changes near the substrate. For certain parameters the aggregates can break under the influence of the surface.
 Please wait while we load your content...
Please wait while we load your content...

