
Comparing alchemical and physical pathway methods for computing the absolute binding free energy of charged ligands†
 *a
Bin W.
Zhang,
b
*a
Bin W.
Zhang,
b
Abstract
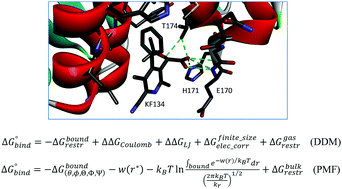
Accurately predicting absolute binding free energies of protein–ligand complexes is important as a fundamental problem in both computational biophysics and pharmaceutical discovery. Calculating binding free energies for charged ligands is generally considered to be challenging because of the strong electrostatic interactions between the ligand and its environment in aqueous solution. In this work, we compare the performance of the potential of mean force (PMF) method and the double decoupling method (DDM) for computing absolute binding free energies for charged ligands. We first clarify an unresolved issue concerning the explicit use of the binding site volume to define the complexed state in DDM together with the use of harmonic restraints. We also provide an alternative derivation for the formula for absolute binding free energy using the PMF approach. We use these formulas to compute the binding free energy of charged ligands at an allosteric site of HIV-1 integrase, which has emerged in recent years as a promising target for developing antiviral therapy. As compared with the experimental results, the absolute binding free energies obtained by using the PMF approach show unsigned errors of 1.5–3.4 kcal mol−1, which are somewhat better than the results from DDM (unsigned errors of 1.6–4.3 kcal mol−1) using the same amount of CPU time. According to the DDM decomposition of the binding free energy, the ligand binding appears to be dominated by nonpolar interactions despite the presence of very large and favorable intermolecular ligand–receptor electrostatic interactions, which are almost completely cancelled out by the equally large free energy cost of desolvation of the charged moiety of the ligands in solution. We discuss the relative strengths of computing absolute binding free energies using the alchemical and physical pathway methods.
 Please wait while we load your content...
Please wait while we load your content...

