
Computational simulations determining disulfonic stilbene derivative bioavailability within human serum albumin†

Abstract
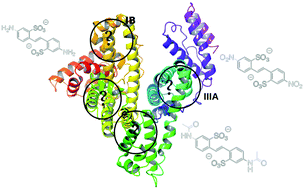
Disulfonic stilbene (DS) derivatives are a member of the large family of compounds widely employed in medicine and biology as modulators for membrane transporters or inhibitors of a protein involved in DNA repair. They constitute interesting compounds that have not yet been investigated within the bioavailability framework. No crystallographic structures exist involving such compounds embedded in the most common drug carrier, human serum albumin (HSA). The present work studies, for the first time, the physico-chemical features driving the inclusion of three DS derivatives (amino, nitro and acetamido, named DADS, DNDS and DATDS, respectively) within the four common HSA binding sites using combined molecular docking and molecular dynamics simulations. A careful analysis of each ligand within each of the studied binding sites is carried out, highlighting specific interactions and key residues playing a role in stabilizing the ligand within each pocket. The comparison between DADS, DNDS and DATDS reveals that depending on the binding site, the conclusions are rather different. For instance, the IB binding site shows a specificity to DADS compounds while IIIA is the most favorable site for DNDS and DATDS.
 Please wait while we load your content...
Please wait while we load your content...

