
Adsorption of oxygen and CO oxidation on Au/anatase(001) catalysts. A DFT+U study

Abstract
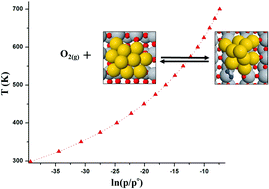
DFT calculations were performed to probe CO oxidation over Au/TiO2 catalysts by adsorbed oxygen. Adsorption dynamics of the O2 molecule like geometric properties, binding energy, electronic structure and dissociation mechanism were investigated. The calculations indicate that adsorption of oxygen is most favorable at the periphery of Au/TiO2 with a binding energy of −139 kJ mol−1. The electronic structure of the adsorbed oxygen indicates that the adsorption is governed by the p–d hybridization of oxygen and Au, with a small contribution from Ti atoms of the support. Upon adsorption of oxygen, the Au cluster loses 0.92 e to adsorbed oxygen. The charge lost by the Au cluster is equally shared by both oxygen atoms (−0.46 e each). Dissociation of oxygen into oxygen adatoms is exothermic and activated by 56 kJ mol−1. Finally, the CO oxidation by adatoms is highly exothermic and activated by an activation barrier of 39.4 kJ mol−1. The calculations show that both oxygen dissociation and CO oxidation are fairly easy on the Au/anatase catalyst.
 Please wait while we load your content...
Please wait while we load your content...

