
Multi-spectroscopic and theoretical analyses on the diphenyl ether–tert-butyl alcohol complex in the electronic ground and electronically excited state†

Abstract
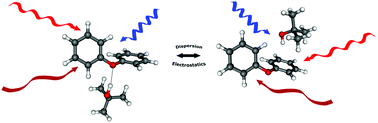
Aromatic ethers such as diphenyl ether (DPE) represent molecules with different docking sites for alcohols leading to competing OH–O and OH–π interactions. In a multi-spectroscopic approach in combination with quantum chemical calculations the complex of DPE with tert-butyl alcohol (t-BuOH) is investigated in the electronic ground state (S0) and the electronically excited state (S1). FTIR, microwave as well as mass- and isomer-selective IR/R2PI spectra are recorded, revealing co-existing OH–O and OH–π isomers in the S0 state. Surprisingly, they are predicted to be of almost equal stability in contrast to the previously investigated DPE–MeOH complex, where the OH–π structure is preferred by both theory and experiment. The tert-butyl group in t-BuOH allows for a simultaneous optimization of hydrogen-bonding and dispersion interactions, which provides a sensitive meeting point between theory and experiment. In the electronically excited state of DPE–t-BuOH, vibrational spectra could be recorded separately for both isomers using UV/IR/UV spectroscopy. In the S1 state the same structural binding motifs are obtained as in the S0 state with the OH–O bond being weakened for the OH–O arrangement and the OH–π interaction being strengthened in the case of the OH–π isomer compared to the S0 state.
 Please wait while we load your content...
Please wait while we load your content...

