
Theoretical study of HCN–water interaction: five dimensional potential energy surfaces

Abstract
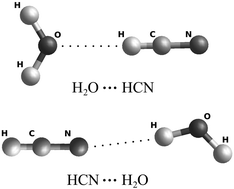
A new five-dimensional potential energy surface is calculated at the coupled-cluster CCSD(T) level of theory for the HCN–water system, treating both monomers as rigid rotors. The associated methodology, which combines extensive ab initio calculations of moderate accuracy (CCSD(T)/AVDZ) and a fitting procedure involving a much lower angular coverage with more accurate ab initio calculations (CCSD(T)/CBS), is described in detail. This methodology provides a time-saving approach to compute quantitatively accurate potential energy surfaces with reasonable computational effort. Our potential reproduces the main features reported in the literature, and will allow us to perform the first quantum and semi-classical simulations of the collisional dynamic on this system.
 Please wait while we load your content...
Please wait while we load your content...

