
Band gap opening in stanene induced by patterned B–N doping†
 *ab
*ab
Abstract
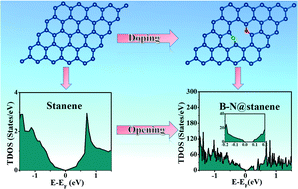
Stanene is a quantum spin Hall insulator and a promising material for electronic and optoelectronic devices. Density functional theory (DFT) calculations are performed to study the band gap opening in stanene by elemental mono-doping (B, N) and co-doping (B–N). Different patterned B–N co-doping is studied to change the electronic properties of stanene. A patterned B–N co-doping opens the band gap in stanene and its semiconducting nature persists under strain. Molecular dynamics (MD) simulations are performed to confirm the thermal stability of such a doped system. The stress–strain study indicates that such a doped system is as stable as pure stanene. Our work function calculations show that stanene and doped stanene have a lower work function than graphene and thus are promising materials for photocatalysts and electronic devices.
 Please wait while we load your content...
Please wait while we load your content...

