
An optimized charge penetration model for use with the AMOEBA force field†

Abstract
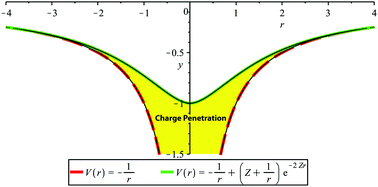
The principal challenge of using classical physics to model biomolecular interactions is capturing the nature of short-range interactions that drive biological processes from nucleic acid base stacking to protein–ligand binding. In particular most classical force fields suffer from an error in their electrostatic models that arises from an ability to account for the overlap between charge distributions occurring when molecules get close to each other, known as charge penetration. In this work we present a simple, physically motivated model for including charge penetration in the AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications) force field. With a function derived from the charge distribution of a hydrogen-like atom and a limited number of parameters, our charge penetration model dramatically improves the description of electrostatics at short range. On a database of 101 biomolecular dimers, the charge penetration model brings the error in the electrostatic interaction energy relative to the ab initio SAPT electrostatic interaction energy from 13.4 kcal mol−1 to 1.3 kcal mol−1. The model is shown not only to be robust and transferable for the AMOEBA model, but also physically meaningful as it universally improves the description of the electrostatic potential around a given molecule.
 Please wait while we load your content...
Please wait while we load your content...

