
A semi-classical approach to the calculation of highly excited rotational energies for asymmetric-top molecules
 a
Per
Jensen
*d
a
Per
Jensen
*d
Abstract
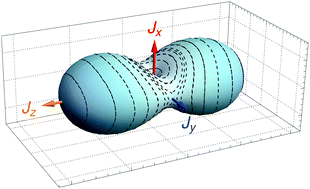
We report a new semi-classical method to compute highly excited rotational energy levels of an asymmetric-top molecule. The method forgoes the idea of a full quantum mechanical treatment of the ro-vibrational motion of the molecule. Instead, it employs a semi-classical Green's function approach to describe the rotational motion, while retaining a quantum mechanical description of the vibrations. Similar approaches have existed for some time, but the method proposed here has two novel features. First, inspired by the path integral method, periodic orbits in the phase space and tunneling paths are naturally obtained by means of molecular symmetry analysis. Second, the rigorous variational method is employed for the first time to describe the molecular vibrations. In addition, we present a new robust approach to generating rotational energy surfaces for vibrationally excited states; this is done in a fully quantum-mechanical, variational manner. The semi-classical approach of the present work is applied to calculating the energies of very highly excited rotational states and it reduces dramatically the computing time as well as the storage and memory requirements when compared to the fullly quantum-mechanical variational approach. Test calculations for excited states of SO2 yield semi-classical energies in very good agreement with the available experimental data and the results of fully quantum-mechanical calculations.
 Please wait while we load your content...
Please wait while we load your content...

