
Axial coordination and electronic structure of diatomic NO, CO, and O2 molecules adsorbed onto Co-tetraphenylporphyrin on Au(111), Ag(111), and Cu(111): a density-functional theory study
Abstract
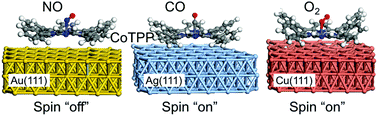
Based on density functional theory calculations, we investigated the axial bindings of diatomic molecules (NO, CO, and O2) to metalloporphyrins and their spin switching behaviors. These binding reactions provide the ways to control molecular states and spins in metalloporphyrin systems that can be used in bio-sensing and spintronic applications. To microscopically understand the non-trivial axial binding structures and spin-switching behaviors of diatomic molecules (NO, CO, and O2) adsorbed onto Co-tetraphenylporphyrin (CoTPP), we performed spin-polarized density functional theory (DFT) calculations for various CoTPP systems on Au(111), Ag(111), and Cu(111) substrates. We also systematically evaluated the effects of van der Waals and Hubbard U corrections by directly comparing the electronic structure with the results of scanning tunnelling microscopy. From the calculation results, we have found that a NO molecule, almost 60° tilted away from the axial direction, is coordinated with CoTPP with a binding energy of 1.2–1.7 eV depending on substrates, and the spin state of CoTPP is completely switched off due to the charge transfer from NO to CoTPP. On the other hand, CO and O2 molecules rather weakly interact with a binding energy of 0.4–0.8 eV, and a spin polarization of ∼1μB is still present at CoTPP. A CO molecule is expected to be almost straightly coordinated along the axial direction of CoTPP, but O2 is tilted similarly to the NO case. Regarding the substrate effects, we have found that there is noticeable charge transfer from Ag(111) and Cu(111) to CoTPP, but no significant charge transfer from Au(111) to CoTPP. These findings of the axial coordination and spin states for NO, CO, and O2 adsorbed CoTPP systems will be useful for understanding bio-sensing mechanisms and designing molecular spintronic systems.
- This article is part of the themed collection: Molecular Spintronics : The role of Coordination Chemistry
 Please wait while we load your content...
Please wait while we load your content...

