
A potential role of a substrate as a base for the deprotonation pathway in Rh-catalysed C–H amination of heteroarenes: DFT insights†
Abstract
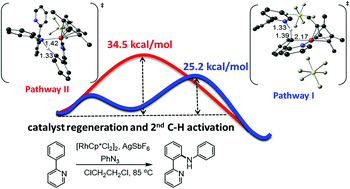
The possibility of direct introduction of a new functionality through C–H bond activation is an attractive strategy in covalent synthesis. Here, we investigated the mechanism of Rh-catalysed C–H amination of the heteroaryl substrate (2-phenylpyridine) using phenyl azide as a nitrogen source by density functional theory (DFT). For the deprotocyclometallation and protodecyclometallation processes of the title reaction, we propose a stepwise base-assisted mechanism (pathway I) instead of the previously reported concerted mechanism (pathway II). In the new mechanism proposed here, 2-phenylpyridine acts as a base in the initial deprotonation step (C–H bond cleavage) and transports the proton towards the final protonation step. In fact, the N–H bond of the strong conjugate acid (formed during the initial C–H bond cleavage) considered in pathway I (viaTS4) is more acidic than the C–H bond of the neutral substrate considered in pathway II (viaTS5). The higher activation barrier of TS5 mainly originates from the ring strain of the four-membered cyclic transition state. The vital role of the base, as disclosed here, can potentially have broader mechanistic implications for the development of reaction conditions of transition metal-catalysed reactions.
 Please wait while we load your content...
Please wait while we load your content...

