
Modelling photophysical properties of metal–organic frameworks: a density functional theory based approach†
 *a
and
*a
and
Abstract
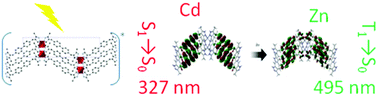
Design of optical properties within metal–organic frameworks (MOFs) is a subject of ever increasing attention in recent years with theoretical approaches poised to play a key role alongside experiment in both the understanding of fundamental mechanisms and the further development of high performance materials. We have developed and applied a simple and computationally affordable protocol rooted in density functional theory (DFT) and its time dependent counterpart (TD-DFT) to two isostructural MOFs based on a 4,4′-bis((3,5-dimethyl-1H-pyrazol-4-yl)methyl)-biphenyl (H2DMPMB) linker. These systems show a remarkable dependence of photoluminescence properties on the interchange of zinc and cadmium cations as building units. Our investigation was able to successfully rationalize the subtle change in the photoluminescence mechanism experimentally observed responsible for the large (0.88 eV) red shift (from 335 nm to 441 nm) observed when going from the cadmium to the zinc based structure. More generally, this computational protocol seems well adapted for the characterization and rationalization of the absorption and emission behaviour of such complex extended materials.
 Please wait while we load your content...
Please wait while we load your content...

