
A DFT and multi-configurational perturbation theory study on O2 binding to a model heme compound via the spin-change barrier†
Abstract
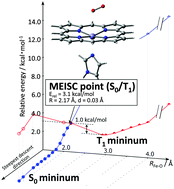
Dioxygen binding to a model heme compound via intersystem crossing (ISC) was investigated with a multi-state multi-configurational self-consistent field method with second-order perturbation theory (MS-CASPT2) and density functional theory (DFT) calculations. In elongated Fe–O distances, the energy levels of the S0 and T1 states are separated, which decreases the probability of intersystem crossing in these structures. At the DFT(B97D) level of calculation, the Fe–O distances of the S0 and T1 states were 1.91 and 2.92 Å, respectively. The minimum energy intersystem crossing point (MEISCP) was located as a transition state at a Fe–O distance of 2.17 Å with an energy barrier of 1.0 kcal mol−1 from the T1 minimum. The result was verified with MS-CASPT2 calculations including the spin–orbit interaction which also showed the intersystem crossing point at a Fe–O distance of 2.05 Å. An energy decomposition analysis on the reaction coordinate showed the important contribution of the ring-shrinking mode of the porphyrin ring, indicating that the reaction coordinates which control the relative energy level of the spin-states play a key role in intersystem crossing.
 Please wait while we load your content...
Please wait while we load your content...

