
Analysis of transition state stabilization by non-covalent interactions in the Houk–List model of organocatalyzed intermolecular Aldol additions using functional-group symmetry-adapted perturbation theory†
Abstract
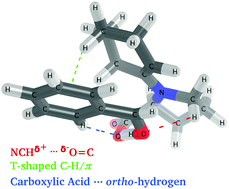
Rational design of catalysts would be aided by a better understanding of how non-covalent interactions stabilize transition states. Here, we apply the newly-developed Functional-Group Symmetry-Adapted Perturbation Theory (F-SAPT) to quantify non-covalent interactions in transition states of the proline-catalyzed intermolecular aldol reaction between benzaldehyde and cyclohexanone, according to the Houk–List mechanism [Bahmanyar et al., J. Am. Chem. Soc., 2003, 125, 2475]. A recent re-examination of this organocatalytic reaction by Rzepa and co-workers [Armstrong et al., Chem. Sci., 2014, 5, 2057] used electron density analysis to identify three key non-covalent interactions thought to influence stereoselectivity: (1) a favorable electrostatic interaction (originally identified by Houk and List) between the NCHδ+ group of the enamine intermediate and the δ−O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of benzaldehyde; (2) a C–H/π interaction between the cyclohexene group of the enamine intermediate and the benzaldehyde phenyl ring; (3) a stabilizing contact between an ortho-hydrogen of the phenyl and an oxygen of the carboxylic acid group of the enamine. These three interactions have been directly computed using F-SAPT, which confirms the stabilizing interaction between an ortho-hydrogen and the carboxylic acid in the (S,S) and (R,S) transition state stereoisomers. F-SAPT analysis also finds stabilizing dispersion and electrostatic interactions due to a C–H/π interaction between the cyclohexene and phenyl groups in the (S,S) and (R,R) transition states. However, unfavorable exchange-repulsion cancels the attractive terms that favor these stereoisomers. Surprisingly, the interaction thought to be most important for stereoselectivity, the NCHδ+⋯δ−OC interaction, is actually found to be repulsive due to the negative charge on the nitrogen. Hence, our results indicate that geometric analysis and/or density-based analysis does not necessarily produce a reliable picture of non-covalent stabilization. As confirmed by high-level coupled-cluster computations, intermolecular interaction energies are strongest for the (R,R) transition states, which are not the experimentally favored products. This suggests that at least for this reaction, stereoselectivity is also strongly dependent on the energy required to distort the reacting molecules into the transition state geometry.
C of benzaldehyde; (2) a C–H/π interaction between the cyclohexene group of the enamine intermediate and the benzaldehyde phenyl ring; (3) a stabilizing contact between an ortho-hydrogen of the phenyl and an oxygen of the carboxylic acid group of the enamine. These three interactions have been directly computed using F-SAPT, which confirms the stabilizing interaction between an ortho-hydrogen and the carboxylic acid in the (S,S) and (R,S) transition state stereoisomers. F-SAPT analysis also finds stabilizing dispersion and electrostatic interactions due to a C–H/π interaction between the cyclohexene and phenyl groups in the (S,S) and (R,R) transition states. However, unfavorable exchange-repulsion cancels the attractive terms that favor these stereoisomers. Surprisingly, the interaction thought to be most important for stereoselectivity, the NCHδ+⋯δ−OC interaction, is actually found to be repulsive due to the negative charge on the nitrogen. Hence, our results indicate that geometric analysis and/or density-based analysis does not necessarily produce a reliable picture of non-covalent stabilization. As confirmed by high-level coupled-cluster computations, intermolecular interaction energies are strongest for the (R,R) transition states, which are not the experimentally favored products. This suggests that at least for this reaction, stereoselectivity is also strongly dependent on the energy required to distort the reacting molecules into the transition state geometry.
 Please wait while we load your content...
Please wait while we load your content...

