
Structure based investigation on the binding interaction of transport proteins in leishmaniasis: insights from molecular simulation†
Abstract
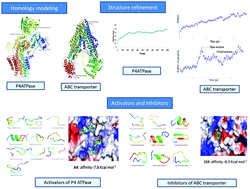
Leishmania major is the causative agent of cutaneous leishmaniasis which affects over 1 million people in 88 different countries. The incidence of this disease is on the rise due to the current problems associated with the present chemotherapeutics. In addition, Leishmania confronts resistance to the traditional drugs like sodium stibogluconate and newer repurposed drugs like miltefosine. ABC transporters are involved in the development of drug resistance. Miltefosine, the drug used for the treatment of leishmaniasis, is effluxed by P4 ATPase and ABC transporter, which is the prime focus of our study in this paper. P4 ATPase (MDR1) along with an unnamed protein (cdc50) translocates miltefosine from the outer to the inner leaflet by the process of flipping which is ATP driven. In contrast, miltefosine also escapes from the cells by an energy dependent mechanism that involves the ABC transporter protein (ABC). It is known that certain genes in the parasite amplify the portions of a gene which encodes ABC transporter and P4 ATPase involved in translocating phospholipids and hence resistance to miltefosine. We observed the ABC and P4 ATPase genes, 39 T-box elements were observed in the ABC transporter protein and three elements were observed in the P4 ATPase gene suggesting its role in transcription regulation. To the best of our knowledge, there are no structural and regulatory reports on these two proteins in L. major. Computational structural biology tools may aid in understanding the interaction of miltefosine with the P4-ATPase–cdc50 complex and the ABC transporter. This can be achieved by modeling the target protein structures, studying the dynamics associated with the different domains of the protein and later using activators and inhibitors to alter the functioning of the protein. Molecular dynamics simulation with a lipid bilayer is performed to investigate the conformational changes and structure–activity relationship. As transporters are difficult to model, the relevant structural motifs and domains may help to understand the allosteric relation with the substrate and the cofactors. The dynamics of a protein molecule ultimately defines the functional mechanism involving excursions of multiple conformational states. To understand these functional mechanisms of transporter proteins, computational modeling and simulations will be carried out with the goal of elucidating the atomistic details of allosteric conformational transitions and propagations during the transport processes. In particular these studies are designed to investigate the critical structural and dynamic elements that determine individual and combined ligand-binding specificities, the interactions among transporters, their coupled-proteins and the associations of transporters within the lipid bilayer. The nature of results from such studies also makes it possible to rationally optimize existing ligands for these proteins and develop some new compounds that can shift the conformational equilibrium of transporters which may aid in functional studies leading to drug discovery.
 Please wait while we load your content...
Please wait while we load your content...