
Quantum and classical dynamics of reactive scattering of H2 from metal surfaces
Abstract
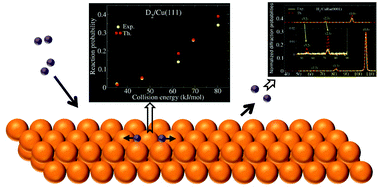
We review the state-of-the art in dynamics calculations on the reactive scattering of H2 from metal surfaces, which is an important model system of an elementary reaction that is relevant to heterogeneous catalysis. In many applications, quantum dynamics and classical trajectory calculations are performed within the Born–Oppenheimer static surface model. However, ab initio molecular dynamics (AIMD) is finding increased use in applications aimed at modeling the effect of surface phonons on the dynamics. Molecular dynamics with electronic friction has been used to model the effect of electron–hole pair excitation. Most applications are still based on potential energy surfaces (PESs) or forces computed with density functional theory (DFT), using a density functional within the generalized gradient approximation to the exchange–correlation energy. A new development is the use of a semi-empirical version of DFT (the specific reaction parameter (SRP) approach to DFT). We also discuss the accurate methods that have become available to represent electronic structure data for the molecule–surface interaction in global PESs. It has now become possible to describe highly activated H2 + metal surface reactions with chemical accuracy using the SRP-DFT approach, as has been shown for H2 + Cu(111) and Cu(100). However, chemical accuracy with SRP-DFT has yet to be demonstrated for weakly activated systems like H2 + Ru(0001) and non-activated systems like H2 + Pd(111), for which SRP DFs are not yet available. There is now considerable evidence that electron–hole pair (ehp) excitation does not need to be modeled to achieve the (chemically) accurate calculation of dissociative chemisorption and scattering probabilities. Dynamics calculations show that phonons can be safely neglected in the chemically accurate calculation of sticking probabilities on cold metal surfaces for activated systems, and in the calculation of a number of other observables. However, there is now sufficient evidence to suggest that the decision on whether or not to neglect phonons should be taken with care, with appropriate consideration of the observable to be computed and of the relevant surface temperature. AIMD calculations have provided valuable insights into the mechanisms that are operative in the dissociative adsorption and absorption of hydrogen on/in precovered metal surfaces. Classical and quantum dynamics calculations have shown that the reaction probability of H2 on Pt surfaces consisting of (100) steps and (111) terraces can to a very good approximation be computed as a weighted average of the reactivities on the steps and terraces. Progress obtained with dynamics calculations on the scattering of H2 from alloys and from simple low index metal surfaces is also reported. Insights that may be obtained on the reactivity of a metal surface from the prominent presence of out-of-plane diffraction or, conversely, the complete absence of diffraction, are discussed. A new field has been opened up by experiments on H2 scattering from surfaces at fast grazing incidence, and we discuss new predictions regarding diffraction and dissociative scattering of H2 under such conditions.
- This article is part of the themed collection: Surface Reaction Dynamics
 Please wait while we load your content...
Please wait while we load your content...

