
Density functional reactivity theory study of SN2 reactions from the information-theoretic perspective
Abstract
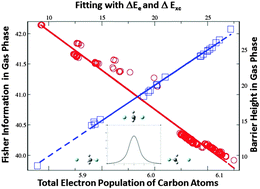
As a continuation of our recent efforts to quantify chemical reactivity with quantities from the information-theoretic approach within the framework of density functional reactivity theory, the effectiveness of applying these quantities to quantify electrophilicity for the bimolecular nucleophilic substitution (SN2) reactions in both gas phase and aqueous solvent is presented in this work. We examined a total of 21 self-exchange SN2 reactions for the compound with the general chemical formula of R1R2R3C–F, where R1, R2, and R3 represent substituting alkyl groups such as –H, –CH3, –C2H5, –C3H7, and –C4H9 in both gas and solvent phases. Our findings confirm that scaling properties for information-theoretic quantities found elsewhere are still valid. It has also been verified that the barrier height has the strongest correlation with the electrostatic interaction, but the contributions from the exchange–correlation and steric effects, though less significant, are indispensable. We additionally unveiled that the barrier height of these SN2 reactions can reliably be predicted not only by the Hirshfeld charge and information gain at the regioselective carbon atom, as previously reported by us for other systems, but also by other information-theoretic descriptors such as Shannon entropy, Fisher information, and Ghosh–Berkowitz–Parr entropy on the same atom. These new findings provide further insights for the better understanding of the factors impacting the chemical reactivity of this vastly important category of chemical transformations.
 Please wait while we load your content...
Please wait while we load your content...

