
Towards an accurate and computationally-efficient modelling of Fe(ii)-based spin crossover materials
Abstract
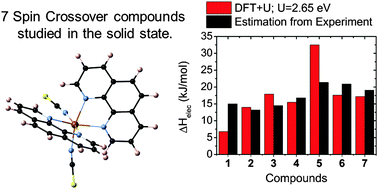
The DFT + U methodology is regarded as one of the most-promising strategies to treat the solid state of molecular materials, as it may provide good energetic accuracy at a moderate computational cost. However, a careful parametrization of the U-term is mandatory since the results may be dramatically affected by the selected value. Herein, we benchmarked the Hubbard-like U-term for seven Fe(II)N6-based pseudo-octahedral spin crossover (SCO) compounds, using as a reference an estimation of the electronic enthalpy difference (ΔHelec) extracted from experimental data (T1/2, ΔS and ΔH). The parametrized U-value obtained for each of those seven compounds ranges from 2.37 eV to 2.97 eV, with an average value of U = 2.65 eV. Interestingly, we have found that this average value can be taken as a good starting point since it leads to an unprecedented mean absolute error (MAE) of only 4.3 kJ mol−1 in the evaluation of ΔHelec for the studied compounds. Moreover, by comparing our results on the solid state and the gas phase of the materials, we quantify the influence of the intermolecular interactions on the relative stability of the HS and LS states, with an average effect of ca. 5 kJ mol−1, whose sign cannot be generalized. Overall, the findings reported in this manuscript pave the way for future studies devoted to understand the crystalline phase of SCO compounds, or the adsorption of individual molecules on organic or metallic surfaces, in which the rational incorporation of the U-term within DFT + U yields the required energetic accuracy that is dramatically missing when using bare-DFT functionals.
 Please wait while we load your content...
Please wait while we load your content...

