
Singlet and triplet excitons and charge polarons in cycloparaphenylenes: a density functional theory study†
Abstract
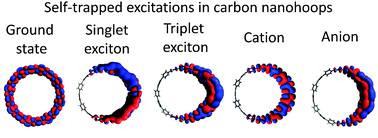
The conformational structure and the electronic properties of various electronic excitations in cycloparaphenylenes (CPPs) are calculated using hybrid density functional theory (DFT). The results demonstrate that wavefunctions of singlet and triplet excitons as well as the positive and negative polarons remain fully delocalized in CPPs. In contrast, these excitations in larger CPP molecules become localized on several phenyl rings, which are locally planarized, while the undeformed ground state geometry is preserved on the rest of the hoop. As evidenced by the measurements of bond-length alternation and dihedral angles, localized regions show stronger hybridization between neighboring bonds and thus enhanced electronic communication. This effect is even more significant in the smaller hoops, where phenyl rings have strong quinoid character in the ground state. Thus, upon excitation, electron–phonon coupling leads to the self-trapping of the electronic wavefunction and release of energy from fractions of an eV up to two eVs, depending on the type of excitation and the size of the hoop. The impact of such localization on electronic and optical properties of CPPs is systematically investigated and compared with the available experimental measurements.
 Please wait while we load your content...
Please wait while we load your content...

