
Theoretical understanding of two-photon-induced fluorescence of isomorphic nucleoside analogs†
Abstract
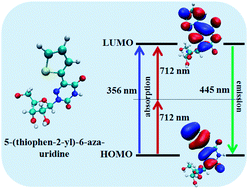
We use ab initio Density Functional Theory (DFT) and Time-dependent DFT (TDDFT) calculations for a detailed understanding of one-photon absorption (1PA) and two-photon absorption (2PA) cross sections of eight different nucleoside analogs. The results are compared and contrasted with the available experimental data. Our calculated results show that the low energy peaks in the absorption spectra mainly arise because of the π–π* electronic transition of the nucleoside analogs. The emission spectra of the nucleoside analogs are also calculated using TDDFT methods. The calculated absorption and emission spectra in the presence of a solvent follow the same trend as those found experimentally. Our results demonstrate that the nucleoside analogs show significantly different electronic and optical properties, although their bonding aspects towards Watson–Crick base pairing remain the same. We also derive the microscopic details of the origin of nonlinear optical properties of the nucleoside analogs.
 Please wait while we load your content...
Please wait while we load your content...

