
Unravelling the impact of hydrocarbon structure on the fumarate addition mechanism – a gas-phase ab initio study†
Abstract
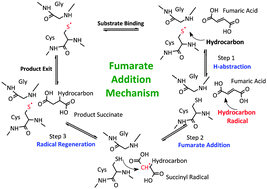
The fumarate addition reaction mechanism is central to the anaerobic biodegradation pathway of various hydrocarbons, both aromatic (e.g., toluene, ethyl benzene) and aliphatic (e.g., n-hexane, dodecane). Succinate synthase enzymes, which belong to the glycyl radical enzyme family, are the main facilitators of these biochemical reactions. The overall catalytic mechanism that converts hydrocarbons to a succinate molecule involves three steps: (1) initial H-abstraction from the hydrocarbon by the radical enzyme, (2) addition of the resulting hydrocarbon radical to fumarate, and (3) hydrogen abstraction by the addition product to regenerate the radical enzyme. Since the biodegradation of hydrocarbon fuels via the fumarate addition mechanism is linked to bio-corrosion, an improved understanding of this reaction is imperative to our efforts of predicting the susceptibility of proposed alternative fuels to biodegradation. An improved understanding of the fuel biodegradation process also has the potential to benefit bioremediation. In this study, we consider model aromatic (toluene) and aliphatic (butane) compounds to evaluate the impact of hydrocarbon structure on the energetics and kinetics of the fumarate addition mechanism by means of high level ab initio gas-phase calculations. We predict that the rate of toluene degradation is ∼100 times faster than butane at 298 K, and that the first abstraction step is kinetically significant for both hydrocarbons, which is consistent with deuterium isotope effect studies on toluene degradation. The detailed computations also show that the predicted stereo-chemical preference of the succinate products for both toluene and butane are due to the differences in the radical addition rate constants for the various isomers. The computational and kinetic modeling work presented here demonstrates the importance of considering pre-reaction and product complexes in order to accurately treat gas phase systems that involve intra and inter-molecular non-covalent interactions.
 Please wait while we load your content...
Please wait while we load your content...

