
Thermal properties of molecular crystals through dispersion-corrected quasi-harmonic ab initio calculations: the case of urea†
Alessandro
Erba,  *a
*a
*a
Abstract
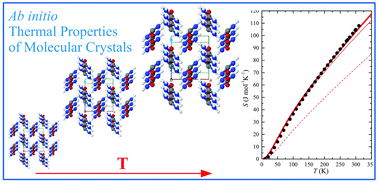
An ab initio quantum-mechanical theoretical framework is presented to compute the thermal properties of molecular crystals. The present strategy combines dispersion-corrected density-functional-theory (DFT-D), harmonic phonon dispersion, quasi-harmonic approximation to the lattice dynamics for thermal expansion and thermodynamic functions, and quasi-static approximation for anisotropic thermo-elasticity. The proposed scheme is shown to reliably describe thermal properties of the urea molecular crystal by a thorough comparison with experimental data.
 Please wait while we load your content...
Please wait while we load your content...

