
Dramatic effect of pore size reduction on the dynamics of hydrogen adsorbed in metal–organic materials
Abstract
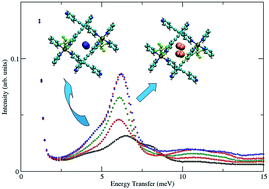
The effects of pore size reduction on the dynamics of hydrogen sorption in metal–organic materials (MOMs) were elucidated by studying SIFSIX-2-Cu and its doubly interpenetrated polymorph SIFSIX-2-Cu-i by means of sorption, inelastic neutron scattering (INS), and computational modeling. SIFSIX-2-Cu-i exhibits much smaller pore sizes, which possess high H2 sorption affinity at low loadings. Experimental H2 sorption measurements revealed that the isosteric heat of adsorption (Qst) for H2 in SIFSIX-2-Cu-i is nearly two times higher than that for SIFSIX-2-Cu (8.6 vs. 4.6 kJ mol−1). The INS spectrum for H2 in SIFSIX-2-Cu-i is rather unique for a porous material, as only one broad peak appears at low energies near 6 meV, which simply increases in intensity with loading until the pores are filled. The value for this rotational transition is lower than that in most neutral metal–organic frameworks (MOFs), including those with open Cu sites (8–9 meV), which is indicative of a higher barrier to rotation and stronger interaction in the channels of SIFSIX-2-Cu-i than the open Cu sites in MOFs. Simulations of H2 sorption in SIFSIX-2-Cu-i revealed two hydrogen sorption sites in the MOM: direct interaction with the equatorial fluorine atom (site 1) and between two equatorial fluorine atoms on opposite walls (site 2). The calculated rotational energy levels and rotational barriers for the two sites in SIFSIX-2-Cu-i are in good agreement with INS data. Furthermore, the rotational barriers and binding energies for site 2 are slightly higher than that for site 1, which is consistent with INS results. The lowest calculated transition for the primary site in SIFSIX-2-Cu is also in good agreement with INS data. In addition, this transition in the non-interpenetrating material is higher than any of the sites in SIFSIX-2-Cu-i, which indicates a significantly weaker interaction with the host as a result of the larger pore size.
 Please wait while we load your content...
Please wait while we load your content...

