
A precision structural model for fullerenols†
Abstract
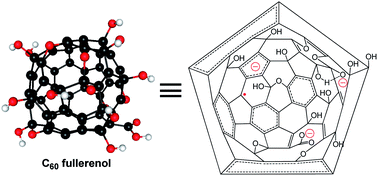
Fullerenol is one of the main precursors of fullerene-based materials, which is promising for various biological applications because of its unusual biocompatibility and biofunctionality. However, the functional groups, acidity and reducibility, which substantialize the applications of fullerenols, remain open questions. Using density functional theory calculations, we investigated reaction mechanisms underlying the acidity and reducibility of C60 fullerenols. On the basis of theoretical insights combined with synthesis, IR and NMR structural characterization of 13C-labeled C60 fullerenols, we identified the functional groups and developed and verified a structural model for C60 fullerenols. The results show a strong dependence of acidity and reducibility on the hydroxyl distributions of fullerenols. Fullerenols with stable π-electron configurations on C60 cores, in which no double bonds have to be placed in conjugated pentagonal rings when drawing their Kekulé structures, have low acidities and reducibilities. Contrarily, fullerenols with unstable π-electron configurations have high acidities and reducibilities. For fullerenols with different hydroxyl distributions, the calculated acid dissociation constants range from −17.55 to 15.21, and the calculated redox potentials range from −0.87 to 1.32 V. Hydroxyls with high acidities and reducibilities unlikely survive in fullerenols synthesized using alkali, oxidizing conditions. Instead, they exist as the corresponding conjugate bases or oxidized products. This structural prediction agrees with our NMR results and the previous experiments. The proposed structural model is able to interpret the main IR and NMR spectroscopic and electronically paramagnetic properties of C60 fullerenols without the disadvantages of the previous models.
 Please wait while we load your content...
Please wait while we load your content...

