
A DFT and ONIOM study of C–H hydroxylation catalyzed by nitrobenzene 1,2-dioxygenase†
Abstract
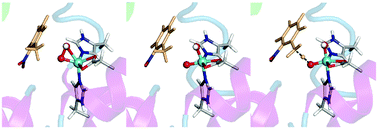
A detailed description of the mechanism of C–H hydroxylation by Rieske non-heme iron dioxygenases remains elusive, as the nature of the oxidizing species is not definitively known. DFT calculations on cluster models of nitrobenzene 1,2-dioxygenase were done to explore possible mechanisms arising from oxidation by either the experimentally observed FeIII–OOH complex or the putative high-valent HO–FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate formed through a heterolytic O–O bond cleavage. Hydrogen abstraction by HO–FeVO, followed by oxygen rebound, was found to be consistent with experimental studies. The findings from the quantum mechanical cluster approach were verified by accounting for the effect of the protein environment on transition state geometries and reaction barriers through ONIOM calculations.
O intermediate formed through a heterolytic O–O bond cleavage. Hydrogen abstraction by HO–FeVO, followed by oxygen rebound, was found to be consistent with experimental studies. The findings from the quantum mechanical cluster approach were verified by accounting for the effect of the protein environment on transition state geometries and reaction barriers through ONIOM calculations.
 Please wait while we load your content...
Please wait while we load your content...

