
Structures and stability of AnO4 isomers, An = Pu, Am, and Cm: a relativistic density functional study
Abstract
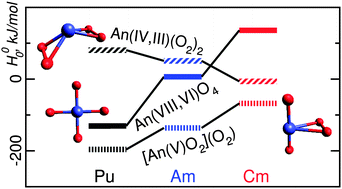
Equilibrium structures and energetics of various isomers of molecules with stoichiometry An·4O (An = Pu, Am, and Cm) are studied through electronic structure calculations at the relativistic density functional theory level in the frame of an accurate small-core pseudopotential model. In all cases, the global minima of the An·4O potential energy surfaces correspond to dioxo-superoxido-like species, [AnO2](O2). The stability of the “true” oxides AnO4 decreases from Pu to Cm, whereas the isomers with two O2 groups become relatively more stable. Correlation between the formal oxidation states and the Bader net charges of actinide atoms is discussed. Structural parameters, vibrational frequencies and charge and spin magnetization density distributions are analyzed in order to characterize the different isomers in chemical terms. Decrease of the An oxidation states along the An series is evident.
 Please wait while we load your content...
Please wait while we load your content...

