
Hydration of the cyanide ion: an ab initio quantum mechanical charge field molecular dynamics study
Abstract
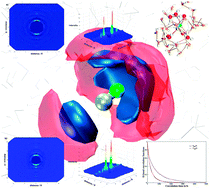
This paper presents an ab initio quantum mechanical charge field molecular dynamics simulation study of the cyanide anion (CN−) in aqueous solution where hydrogen bond formation plays a dominant role in the hydration process. Preferential orientation of water hydrogens compared to oxygen atoms was quantified in terms of radial, angular as well as coordination number distributions. All structural results indicate that the water hydrogens are attracted towards CN− atoms, thus contributing to the formation of the hydration layer. Moreover, a clear picture of the local arrangement of water molecules around the ellipsoidal CN− ion is provided via angular-radial distribution and spatial distribution functions. Apart from the structural analysis, the evaluation of water dynamics in terms of ligand mean residence times and H-bond correlation functions indicates the weak structure making capacity of the CN− ion. The similar values of H-bond lifetimes obtained for the N⋯Hwat and C⋯Hwat bonds indicate an isokinetic behaviour of these H-bonds, since there is a very small difference in the magnitude of the lifetimes. On the other hand, the H-bond lifetimes between water molecules of the hydration shell, and between solute and solvent evidence the slightly stable hydration of the CN−. Overall, the H-bonding dominates in the hydration process of the cyanide anion enabling it to become soluble in the aqueous environment associated to chemical and biological processes.
 Please wait while we load your content...
Please wait while we load your content...

