
Geometric and electronic properties of graphene modified by “external” N-containing groups
Abstract
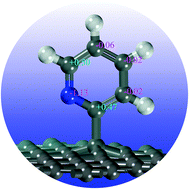
Using first-principles spin polarized density functional theory (DFT) calculations, we investigated structures and electronic properties of “external” nitrogen-containing group (pyridine derivatives) modified graphene via a single or a double bonding mode. Our results show that in the most stable structures, the bonding between pyridine derivatives and graphene involves the ortho-carbon of pyridine derivatives, as confirmed by the Bader charge analysis. The enhanced stability of pyridine derivatives on graphene by [2+2] cycloaddition, e.g., a double bonding mode (DBPyNG), is caused by the matches between frontier orbitals of pyridine derivatives and those of graphene, which leads to the formation of stronger chemical bonds. Interestingly, electronic structure density of states (DOS) analysis of SBPyNG reveals that the spin-up and spin-down parts are clearly split while it is not the case for the double bonding pyridine derivative modified graphene (DBPyNG).
 Please wait while we load your content...
Please wait while we load your content...

