
Atomistic simulations of TeO2-based glasses: interatomic potentials and molecular dynamics
Abstract
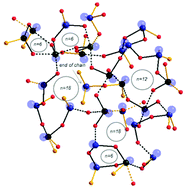
In this work we present for the first time empirical interatomic potentials that are able to reproduce TeO2-based systems. Using these potentials in classical molecular dynamics simulations, we obtained first results for the pure TeO2 glass structure model. The calculated pair distribution function is in good agreement with the experimental one, which indicates a realistic glass structure model. We investigated the short- and medium-range TeO2 glass structures. The local environment of the Te atom strongly varies, so that the glass structure model has a broad Qnm polyhedral distribution. The glass network is described as weakly connected with a large number of terminal oxygen atoms.
 Please wait while we load your content...
Please wait while we load your content...

