
Reactive simulations of the activation barrier to dissolution of amorphous silica in water
Abstract
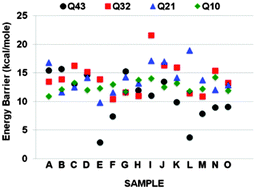
Molecular dynamics simulations employing reactive potentials were used to determine the activation barriers to the dissolution of the amorphous SiO2 surface in the presence of a 2 nm overlayer of water. The potential of mean force calculations of the reactions of water molecules with 15 different starting Q4 sites (Qi is the Si site with i bridging oxygen neighbors) to eventually form the dissolved Q0 site were used to obtain the barriers. Activation barriers for each step in the dissolution process, from the Q4 to Q3 to Q2 to Q1 to Q0 were obtained. Relaxation runs between each reaction step enabled redistribution of the water above the surface in response to the new Qi site configuration. The rate-limiting step observed in the simulations was in both the Q32 reaction (a Q3 site changing to a Q2 site) and the Q21 reaction, each with an average barrier of ∼14.1 kcal mol−1. However, the barrier for the overall reaction from the Q4 site to a Q0 site, averaged over the maximum barrier for each of the 15 samples, was 15.1 kcal mol−1. This result is within the lower end of the experimental data, which varies from 14–24 kcal mol−1, while ab initio calculations using small cluster models obtain values that vary from 18–39 kcal mol−1. Constraints between the oxygen bridges from the Si site and the connecting silica structure, the presence of pre-reaction strained siloxane bonds, and the location of the reacting Si site within slight concave surface contours all affected the overall activation barriers.
 Please wait while we load your content...
Please wait while we load your content...

