
Modelling excited states of weakly bound complexes with density functional theory
Abstract
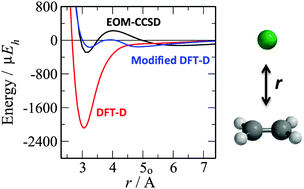
The binding within the ethene–argon and formaldehyde–methane complexes in the ground and electronically excited states is studied with equation of motion coupled cluster theory (EOM-CCSD), second-order Møller–Plesset perturbation theory (MP2) and density functional theory with dispersion corrections (DFT-D). Electronically excited states are studied within MP2 and Kohn–Sham DFT formalisms by exploiting a procedure called the maximum overlap method that allows convergence of the relevant self-consistent field equations to higher energy (or excited state) solutions. Potential energy curves computed using MP2 are in good agreement with the EOM-CCSD calculations for both the valence and Rydberg excited states studied. For the DFT-D approach, B3LYP-D3/aug-cc-pVTZ calculations are found to be in agreement with EOM-CCSD for the ground and valence excited states. However, for the π3s Rydberg state of ethene–argon and the n3s Rydberg state of formaldehyde–methane significant deviation is observed, and this disagreement with EOM-CCSD is present for a variety of DFT-D based approaches. Variation of the parameters within the D2 dispersion correction results in closer agreement with EOM-CCSD for the Rydberg states but demonstrates that a different parameterisation from the ground state is required for these states. This indicates that time-dependent density functional theory calculations based upon a DFT-D reference may be satisfactory for excitations to valence states, but will potentially be inaccurate for excitations to Rydberg states, or more generally states where the nature of the electron density is significantly different from the ground state.
- This article is part of the themed collection: Density functional theory and its applications
 Please wait while we load your content...
Please wait while we load your content...

