
Interplay between molecule–molecule and molecule–substrate interactions: first-principles study of fluoroform aggregates on a hexagonal ice (0001) surface†
Abstract
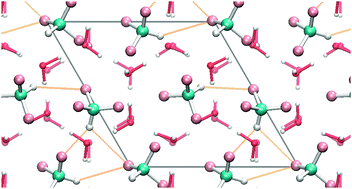
The adsorption of fluoroform molecules on a hexagonal ice (0001) surface was studied using static density functional theory (DFT) calculations and Car–Parrinello molecular dynamics (CP-MD) simulations. Extending our previous work on isolated molecules we focus in the present study on the interplay between molecule–molecule and molecule–substrate interactions. Coverages of up to a full monolayer were modeled by introducing two, three and four fluoroform molecules per unit cell of the ice (0001) substrate. Lowest-energy structures of fluoroform aggregates on the ice surface were determined in a systematic search by performing geometry optimizations from a large set of initial configurations chosen by chemical intuition and from snapshots taken from CP-MD simulations. In the vibrational analysis of the optimized geometries both conventional red- and unusual blue-shifting hydrogen bonds were found. The finite temperature stability of the lowest-energy configurations was probed by CP-MD simulations and conformational changes were analyzed in terms of transformations between the global and local minima structures.
 Please wait while we load your content...
Please wait while we load your content...

