
Identification of non-classical C–H⋯M interactions in early and late transition metal complexes containing the CH(ArO)3 ligand†
Abstract
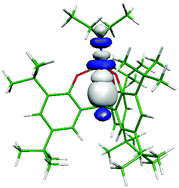
The fully optimised DFT structure of the d0 complex [{CH(ArO)3}Ti(NEt2)] (2) at the B3LYP level compares well with the distorted tetrahedral geometry shown by the X-ray crystal structure. QTAIM analysis of the electron density associated with the C–H⋯Ti interaction shows a well defined bond critical point, a bond path between the hydrogen and titanium centres and a negative value for the energy density indicative of covalency. A natural bond orbital (NBO) picture of the interaction shows that the C–H σ bond electron density donates to a d hybrid orbital on the metal in a linear fashion. Calculated IR and
 Please wait while we load your content...
Please wait while we load your content...

