
Active site of the solvated thiosulfate ion characterized by hydration structures and dynamics
Abstract
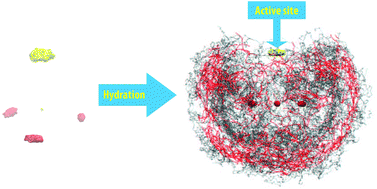
The reactivity of the terminated sulfur atom within the thiosulfate ion (S2O32−) when it is involved in chemical reactions was investigated through the properties of the molecular hydration shell, obtained from the ab initio quantum mechanical change field molecular dynamics (QMCF MD) simulation. The average geometry indicated the significant effect of explicit water on the reduction of the S–S length, which was reflected in the splitting peaks of the spectrum for the stretching mode of this bond (ν(SS)). A further investigation on a simple model with various theoretical levels exhibited the hydrophobicity of the S–S bond. The evaluation of the molecular coordination number was sensitive to the radii of the atomic hydration spheres, which were obtained from the vague boundaries of the first peak in the atomic radial distribution functions. The number of actual contacts specified 6.8 water molecules interacting with the thiosulfate ion, and 2.4 extra waters located in the molecular hydration shell, forming a H-bonding network with the bulk water. The mean residence times for the water ligands distinguished the asymmetric strength of the hydration shell into a weaker sulfur and three stronger oxygen sites, instigating the terminated sulfur atom as the active site that is involved in chemical reactions.
 Please wait while we load your content...
Please wait while we load your content...

