
Quantification of intramolecular cooperativity in polynuclear spin crossover Fe(ii) complexes by density functional theory calculations†
Abstract
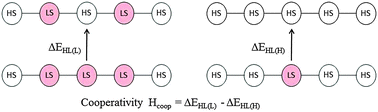
The influence of the spin state of nearest neighbours on the spin transition of a given centre has been modelled with density functional theory methods for the linear oligomeric Fe(II) complexes of
 Please wait while we load your content...
Please wait while we load your content...

