
Charge carrier exchange at chemically modified graphene edges: a density functional theory study†
Abstract
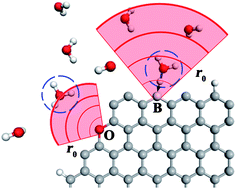
Heteroatom doping on the edge of graphene may serve as an effective way to tune chemical activity of carbon-based electrodes with respect to charge carrier transfer in an aqueous environment. In a step towards developing mechanistic understanding of this phenomenon, we explore herein mechanisms of proton transfer from aqueous solution to pristine and doped graphene edges utilizing density functional theory. Atomic B-, N-, and O- doped edges as well as the native graphene are examined, displaying varying proton affinities and effective interaction ranges with the H3O+ charge carrier. Our study shows that the doped edges characterized by more dispersive orbitals, namely boron and nitrogen, demonstrate more energetically favourable charge carrier exchange compared with oxygen, which features more localized orbitals. Extended calculations are carried out to examine proton transfer from the hydronium ion in the presence of explicit water, with results indicating that the basic mechanistic features of the simpler model are unchanged.
 Please wait while we load your content...
Please wait while we load your content...