
Is gluconate a good model for isosaccharinate in uranyl(vi) chemistry? A DFT study†
Abstract
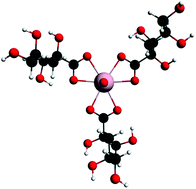
The geometries, relative energies and spectroscopic properties of a range of α-isosaccharinate complexes of uranyl(VI) are studied computationally using ground state and time-dependent density functional theory. The effect of pH is accommodated by varying the number of water and hydroxide ligands accompanying isosaccharinate in the equatorial plane of the uranyl unit. For 1 : 1 complexes, the calculated uranyl ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) asym stretching frequency decreases as pH increases, in agreement with previous experimental data. Three different isosaccharinate chelating modes are studied. Their relative energies are found to be pH dependent, although the energetic differences between them are not sufficient to exclude the possibility of multiple speciation. At higher pH, the uranyl–ligand interactions are dominated more by the equatorial OH− than by the organic ligands. Calculated electronic excitation energies support experiment in finding the lowest energy transitions to be ligand → metal charge transfer. 13C NMR chemical shifts are calculated for the coordinated isosaccharinate in the high pH mimics, and show good agreement with experimental data, supporting the experimental conclusion that the five-membered chelate ring is favoured at high pH. The effect of increasing the isosaccharinate concentration is modelled by calculating 1 : 2 and 1 : 3 uranyl : α-isosaccharinate complexes. Comparison of the results of the present study with those from our closely related investigation of uranyl(VI)–D-gluconate complexes (Dalton Transactions40 (2011) 11248) reveals strong similarities in structure, bonding, coordination geometry and electronic excitations, but also differences in ΔG for key ligand replacement reactions, suggesting that caution should be exercised when using gluconate as a thermodynamic model for isosaccharinate in uranyl(VI) chemistry.
asym stretching frequency decreases as pH increases, in agreement with previous experimental data. Three different isosaccharinate chelating modes are studied. Their relative energies are found to be pH dependent, although the energetic differences between them are not sufficient to exclude the possibility of multiple speciation. At higher pH, the uranyl–ligand interactions are dominated more by the equatorial OH− than by the organic ligands. Calculated electronic excitation energies support experiment in finding the lowest energy transitions to be ligand → metal charge transfer. 13C NMR chemical shifts are calculated for the coordinated isosaccharinate in the high pH mimics, and show good agreement with experimental data, supporting the experimental conclusion that the five-membered chelate ring is favoured at high pH. The effect of increasing the isosaccharinate concentration is modelled by calculating 1 : 2 and 1 : 3 uranyl : α-isosaccharinate complexes. Comparison of the results of the present study with those from our closely related investigation of uranyl(VI)–D-gluconate complexes (Dalton Transactions40 (2011) 11248) reveals strong similarities in structure, bonding, coordination geometry and electronic excitations, but also differences in ΔG for key ligand replacement reactions, suggesting that caution should be exercised when using gluconate as a thermodynamic model for isosaccharinate in uranyl(VI) chemistry.
 Please wait while we load your content...
Please wait while we load your content...

