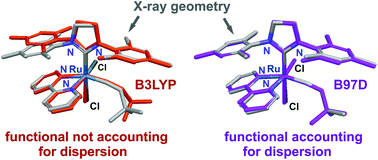
We have investigated the performance of eight popular density functionals, four of which are “standard” functionals not including dispersion (B3LYP, BP86, PBE, and TPSS) and four of which have been constructed to account for dispersion (B97D, wB97XD, M06, and M06L), in reproducing 18 molecular structures derived from single-crystal X-ray diffraction experiments on ruthenium-based olefin metathesis catalyst precursors. Our analysis of all the internuclear distances reveals that standard DFT predicts systematically expanded structures. In contrast, all the methods accounting for dispersion give rise to more compact structures, removing the systematic overestimation of internuclear distances. The contracting effect of dispersion is general and also affects chemical bonds, thus reducing the general overestimation of bond lengths. The best overall performance is observed for wB97XD, which offers relatively small statistical errors when considering the overall structure as well as selected distances. Only for the coordination center geometry is the accuracy of wB97XD matched by standard functionals such as PBE and TPSS, whereas M06 and M06L are associated with larger errors. At the other end of the scale, B3LYP is seen to give the largest statistical errors in general, both when considering the complete structures and the geometries of the coordination centers alone. For the organic ligands, however, B3LYP performs clearly better than the other standard functionals although not as well as the functionals accounting for dispersion. Extending the basis sets is seen to improve the structures in particular of the coordination center, thus underlining the importance of using sufficiently flexible basis sets if highly accurate geometries are to be obtained. Similar conclusions to those obtained for the ruthenium catalysts can be drawn from comparisons of the X-ray crystal structures of 10 other organometallic complexes of relevance to homogeneous catalysis, covering first (Ti, Fe, Co, Ni), second (Zr, Mo, Rh, Pd) and third (W, Ir) row transition metals, with those of DFT. The latter analyses thus offer a first indication that the picture obtained for the ruthenium alkylidene complexes may be extended to other classes of relatively large transition metal complexes.

 Please wait while we load your content...
Please wait while we load your content...

