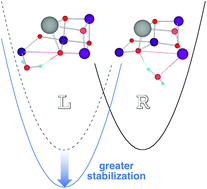
Full geometry optimizations of several inorganic model clusters, CaMn4O4XYZ(H2O)2 (X, Y, Z = H2O, OH− or O2−), by the use of the B3LYP hybrid density functional theory (DFT) have been performed to illuminate plausible molecular structures of the catalytic site for water oxidation in the S0, S1, S2 and S3 states of the Kok cycle for the oxygen-evolving complex (OEC) of photosystem II (PSII). Optimized geometries obtained by the energy gradient method have revealed the degree of symmetry breaking of the unstable three-center Mna–X–Mnd bond in CaMn4O4XYZ(H2O)2. The right-elongated (R) Mna–X⋯Mnd and left-elongated (L) Mna⋯X–Mnd structures appear to occupy local minima on a double-well potential for several key intermediates in these states. The effects of insertion of one extra water molecule to the vacant coordination site, Mnd (Mna), for R (L) structures have also been examined in detail. The greater stability of the L-type structure over the R-type has been concluded for key intermediates in the S2 and S3 states. Implications of the present DFT structures are discussed in relation to previous DFT and related results, together with recent X-ray diffraction results for model compounds of cubane-like OEC cluster of PSII.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


