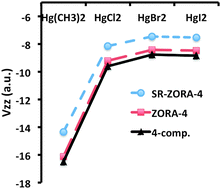
We examine the performance of Density Functional Theory (DFT) approaches based on the Zeroth-Order Regular Approximation (ZORA) Hamiltonian (with and without inclusion of spin–orbit coupling) for predictions of electric field gradients (EFGs) at the heavy atom Hg nucleus. This is achieved by comparing with benchmark DFT and CCSD-T data (Arcisauskaite et al., Phys. Chem. Chem. Phys., 2012, 14, 2651–2657) obtained from 4-component Dirac–Coulomb Hamiltonian calculations. The investigated set of molecules comprises linear HgL2 (L = Cl, Br, I, CH3) and bent HgCl2 mercury compounds as well as the trigonal planar [HgCl3]− system. In 4-component calculations we used the dyall.cv3z basis set for Hg, Br, I and the cc-pCVTZ basis set for H, C, Cl, whereas in ZORA calculations we used the QZ4P basis set for all the atoms. ZORA-4 reproduces the fully relativistic 4-component DFT reference values within 6% for all studied Hg compounds and employed functionals (BH&H, BP86, PBE0), whereas scalar relativistic (SR)-ZORA-4 results show deviations of up to 15%. Compared to our 4-component CCSD-T benchmark the BH&H functional performs best at both 4-component and ZORA levels. We furthermore observe that changes in the largest component of the diagonalised EFG tensor, Vzz, of linear HgCl2 show a slightly stronger dependence than the r−3 scaling upon bond length r(Hg–Cl) alterations. The 4-component/BH&H Vzz value of −9.26 a.u. for a bent HgCl2 (∠Cl–Hg–Cl = 120°) is close to −9.60 a.u. obtained for the linear HgCl2 structure. Thus a point charge model for EFG calculations completely fails in this case. By means of a projection analysis of molecular orbital (MO) contributions to Vzz in terms of the atomic constituents, we conclude that this is due to the increased importance of the Hg 5d orbitals upon bending HgCl2 compared to the linear HgCl2 structure. Changing ligand leads to only minor changes in Vzz (from −9.60 a.u. (HgCl2) to −8.85 a.u. (HgI2) at the 4-component/BH&H level). This appears to be due to cancellation of contributions with opposite signs to Vzz arising from: (i) increasing electron donation from occupied ligand orbitals to the formally empty Hg 6p orbitals and (ii) an increasing bond length and a decreasing negative charge on the ligand along the series.

 Please wait while we load your content...
Please wait while we load your content...

