
Spin crossover transition of Fe(phen)2(NCS)2: periodic dispersion-corrected density-functional study
Abstract
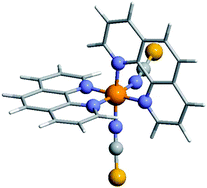
Periodic dispersion corrected DFT calculations have been performed to study the spin-crossover transition of Fe(phen)2(NCS)2 in the molecular and in the crystalline state. We show that London dispersion interactions play a crucial role in the cohesion of the crystals. Based on calculations of vibrational eigenstates of the isolated molecule and of the crystalline phase in both the low- and high-spin states, the transition entropies and enthalpies have been calculated. We demonstrate that, due to the stabilization of the low-spin state by intermolecular dispersion forces, the transition enthalpy at the transition temperature is larger for the crystalline phase in comparison with an isolated molecule. The effective coordination number of the
 Please wait while we load your content...
Please wait while we load your content...

