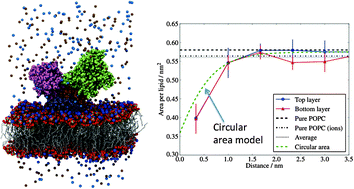
We report large scale molecular dynamics computer simulations, ∼100 ns, of the ion pump Ca2+-ATPase immersed in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer. The structure simulated here, E1, one of the several conformations resolved using X-ray diffraction techniques, hosts two Ca2+-ions in the hydrophobic domain. Our results indicate that protonated residues lead to stronger ion–residue interactions, supporting previous conclusions regarding the sensitivity of the Ca2+ behaviour to the protonated state of the amino acid binding sites. We also investigate how the protein perturbs the bilayer structure. We show that the POPC bilayer is ∼12% thinner than the pure bilayer, near the protein surface. This perturbation decays exponentially with the distance from the protein with a characteristic decay length of 0.8 nm. We find that the projected area per lipid also decreases near the protein. Using an analytical model we show that this change in the area is only apparent and it can be explained by considering the local curvature of the membrane. Our results indicate that the real area per lipid near the protein is not significantly modified with respect to the pure bilayer result. Further our results indicate that the local deformation of the membrane around the protein might be compatible with the enhanced protein activity observed in experiments over a narrow range of membrane thicknesses.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


